Genética del cáncer
|
Traducido de:
Horwitz's Cancer Genetics Lectures
HuBio 554 Medical Genetics, Fall 1997
Dr. Marshall Horwitz, Univ. Washington
Reproducido con permiso del autor |

Nota: los términos médicos pueden no estar
correctamente traducidos
Hay tres tipos de genes relacionados con el cáncer:
1. Oncogenes
Historia
-
1911 Rous: virus del sarcoma de pollo; premio Nobel en 1966.
-
1970 Temin y Baltimore: transcriptasa inversa; premio Nobel en 1975.
-
1976 Bishop y Varmus: oncogenes retrovirales como agentes causales de la
transformación celular; premio Nobel en 1989.
-
1979 Weinberg: descubre por transfección los oncogenes celulares.
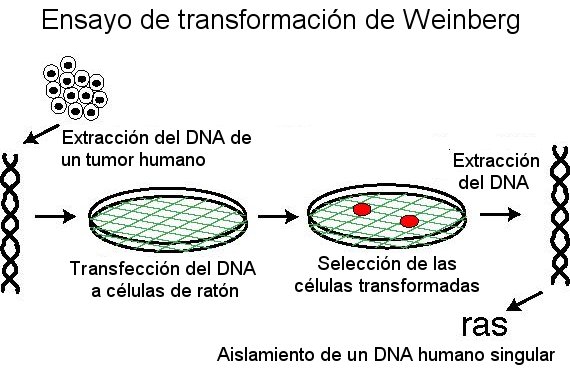
Oncogenes de retrovirus
Un retrovirus es un virus con un gen de RNA que contienen la transcriptasa
inversa para copiar RNA a DNA
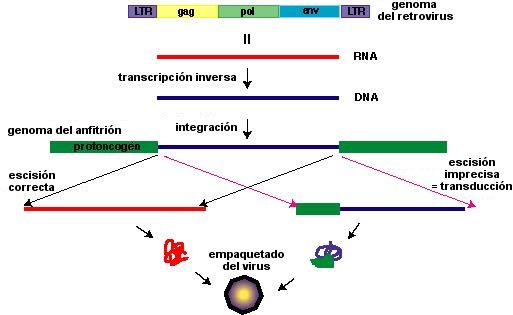
Los oncogenes de los retrovirus son diferentes de los virus transformantes
de DNA (p.ej., el virus del papiloma, causante del cáncer cervical),
cuyos "oncogenes" no tienen homólogos celulares.
Los retrovirus no tienen papel en los tumores humanos (excepto los linfomas
no hodgkininanos y el sarcoma de Kaposi relacionados con el VIH, el HTLV I y
la leucemia de células T en Japón, y el HTLV II en algunas
tricoleucemias (hairy cell leukemias), pero han permitido la identificación
de homólogos de los protoncogenes que están mutados somáticamente
en muchas enfermedades.
Ejemplos de oncogenes
Se conocen alrededor de 50 oncogenes. Los siguientes son bien conocidos:
| Oncogén |
Virus |
Proteína |
| src |
virus del sarcoma de pollo |
proteína tirosina quinasa |
| erbB |
virus de eritroblastosis aviar |
receptor del factor epidérmico de crecimiento (EGF)
/ proteína tirosina quinasa |
| erbA |
virus de eritroblastosis aviar |
receptor de hormonas tiroideas |
| ras |
virus del sarcoma murino |
GTPasa |
| sis |
virus del sarcoma felino |
factor de crecimiento derivado de plaquetas (PDGF) |
| fos |
virus del osteosarcoma murino |
junto a Jun forma el factor de transcripción AP1 |
| jun |
virus del sarcoma aviar |
junto a Fos forma el factor de transcripción AP1 |
| myc |
virus de mielocitomatosis aviar |
factor de transcripción |
La mayoría de los productos de oncogenes se sitúan en
una ruta que regula el crecimiento celular.
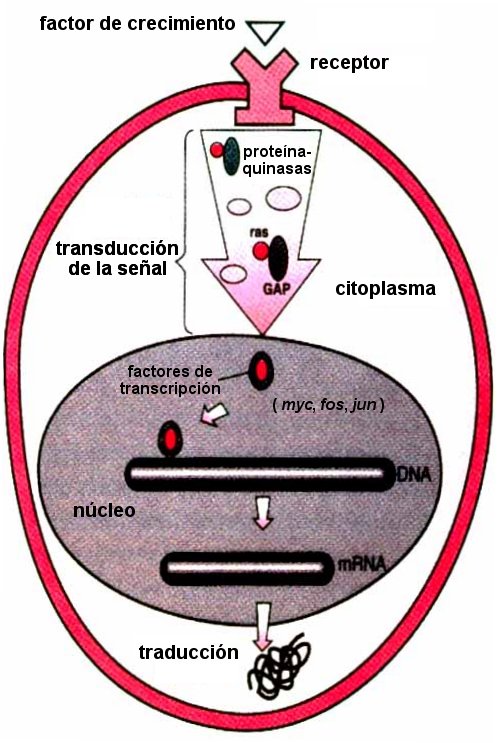
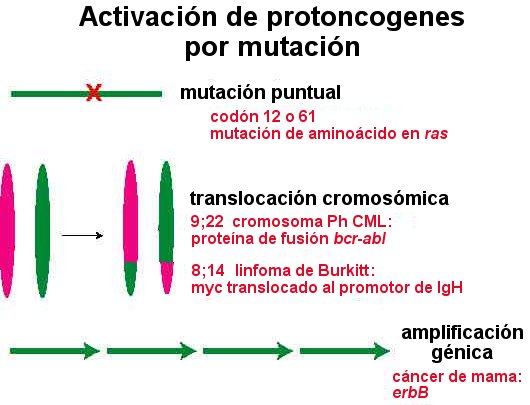
Activación de los protoncogenes
Los protoncogenes se activan por cambios en la secuencia del DNA o
por sobreexpresión; por ejemplo:
Oncogén RET
Se trata del único ejemplo conocido de herencia en la línea germinal
de una mutación en un protoncogén.
Síndrome autosómico dominante MEN2
de carcinoma de la médula tiroidea y feocromocitoma.
 información
información
RET codifica un receptor tirosina quinasa.
La deleción de RET causa una forma autosómica dominante de
megacolon congénito o enfermedad de Hirschsprung.
información
Resumen de oncogenes:
- Identificados como los genes transformantes de los retrovirus.
- Una forma activada de un gen celular (protoncogén).
- Dominantes a nivel celular, lo que significa que basta la mutación
de un alelo.
- Las mutaciones son somáticas y nunca se heredan (excepto
para RET).
- Los retrovirus provocan cáncer en animales, pero no son
una causa significativa de cáncer humano.
- Son reguladores positivos del crecimiento celular.
2. Genes oncosupresores (genes supresores de tumores)
La mayoría de los genes oncosupresores se identificaron a
través de síndromes dominantes autosómicos, poco frecuentes,
de la "familia del cáncer". Posteriormente se ha encontrado que
los mismos genes que están mutados en la línea germinal de esos escasos individuos
son a menudo los que están mutados somáticamente (es decir, en el tumor)
en los casos esporádicos (es decir, no heredados)
del mismo tipo de tumor. Se conocen numerosos síndromes de la familia
del cáncer; aquí se discuten unos pocos de ellos, ejemplos
típicos.
Gen del retinoblastoma
información
Es el prototipo de gen oncosupresor.
~40% de línea germinal, bilateral, inicio infantil
~60% esporádico, unilateral, inicio más tardío
Modelo de dos impactos
Este modelo fue formulado por Knudson en 1971 (two-hit model). El siguiente análisis pretende dar una impresión intuitiva
del razonamiento del modelo cinético de dos impactos, pero carece
del rigor estadístico —y la belleza— del trabajo original de
Knudson.
Una mutación se hereda en la línea germinal y el segundo
impacto es somático.
106 retinoblastos
La tasa de mutación es de alrededor de uno por cada 106 mitosis, implicando
una alta probabilidad de que tendrá lugar el segundo impacto en
aquellos que hereden la primera mutación,
pero la probabilidad de dos impactos somáticos es ~ (10-6)2
x 106 retinoblastos =10-6
(compárese con la incidencia real de 5 x 10-5)
~10% de los casos hereditarios tienen una deleción constitutiva
de al menos parte del cromosoma 13q14.
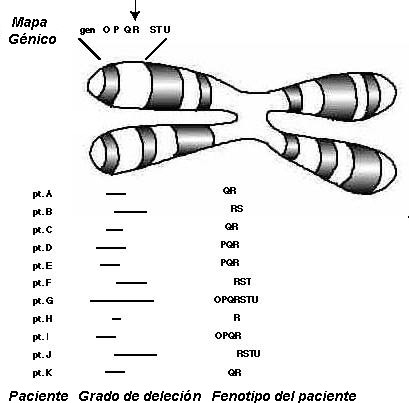
Por tanto, pueden tener un "síndrome de gen contiguo".
Esto ocurre cuando hay una deleción pequeña que abarca al
gen causante de la enfermedad y también a los genes que lo
flanquean. Cada paciente en particular con un síndrome de gen contiguo
para una enfermedad concreta tendrá esa enfermedad con problemas
asociados peculiares (definidos por el grado de deleción de los
genes vecinos y por los genes implicados). Colectivamente, sin embargo,
el grupo de pacientes tendrá un tipo de problemas bastante común,
con una variablidad individual definida de nuevo por el grado de deleción
de los flancos en cada paciente. De modo similar a la mayoría del
resto de pacientes con anomalías cromosómicas estructurales,
aquéllos con síndromes de deleción de genes contiguos
tienen típicamente anomalías congénitas múltiples
(aunque un grupo característicamente particular), retraso mental
y retraso de crecimiento.
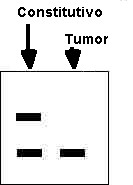
"Carencia de heterocigosis": La ausencia, en el DNA del tumor,
de un polimorfismo presente en el DNA constitutivo. Se toma como evidencia
de una deleción (o un suceso de "conversión " génica)
de un segmento concreto de DNA en un tumor y sugiere la implicación
de un gen supresor de tumores. La carencia de heterocigosis se observa
en los loci supresores de tumores en el DNA extraído de tumores
que aparecen esporádicamente en individuos sin predisposición
familiar y también en tumores de individuos afectados con síndromes
familiares de cáncer. En el último caso, siempre se pierde
el marcador correspondiente al alelo normal.
Los casos heredados de retinoblastoma tienen un riesgo 400 veces superior
de tumores de mesénquima, tales como sarcoma osteogénico,
fibrosarcoma y melanoma. Hay un aumento aún mayor con la radiación
terapéutica del hueso y tejidos blandos de la órbita, pues
la radiación puede inducir el segundo impacto.
RB es una fosfoproteína que regula el paso por el ciclo celular,
uniéndose a factores de transcripción.
Síndrome de Li-Fraumeni y p53
información
-
p53 (17p13): Regula el ciclo celular, es una proteína que se une
al DNA, su expresión está inducida por el daño del
DNA. Se encuentra mutado en numerosos tumores.
-
Síndrome de Li-Fraumeni: herencia autosómica dominante de muchas enfermedades, especialmente
cáncer de mama, sarcoma de tejidos blandos, osteosarcoma, tumores
cerebrales, leucemia y carcinoma adrenocortical. Incidencia: 2 x 10-5.
Posteriormente se ha identificado p53 como el gen responsable.
-
Al igual que RB, p53 está mutado somáticamente en muchas
enfermedades, especialmente cáncer de colon y cáncer pulmonar
de células pequeñas, que no son característicos del
síndrome de Li-Fraumeni.
Neurofibromatosis 1 (NF1)
información
-
Autosómica dominante.
- Incidencia: 3 x 10-4.
-
Neurofibroma benigno, ocasionalmente neurofibrosarcoma maligno, schwanoma,
glioma, feocromocitoma, leucemia.
-
Lesiones de la piel con manchas de color café con leche.
-
Neurofibromina (17q11), GAP (proteína activante de GTPasa): la pérdida
de su actividad produce fallo en la hidrólisis de GTP a GDP.
Síndrome de Von Hippel - Lindau (hemangiomatosis cerebelorretiniana)
información
-
Autosómica dominante.
- Incidencia: 3 x 10-5.
-
Hemangioblastoma del cerebro, feocromocitoma, cáncer de células
renales.
-
3p25, molécula de superficie celular implicada en adhesión
celular y transducción de señales.
-
Carencia de heterocigosis frecuente en el cáncer esporádico
de células renales.
Poliposis adenomatosa familiar
información
-
~1% de todos los cánceres de colon.
-
Los pólipos empiezan en la primera década de vida.
-
APC (5q21), proteína citoplásmica de función desconocida.
-
Mutada también en el adenocarcinoma esporádico de colon,
pero el cáncer de colon es (como probablemente todos los tumores)
el resultado de una ruta de varias etapas que implica a otros genes: ras,
p53, etc.
Cáncer de mama familiar
- Se heredan de forma autosómica dominante entre el 5 y el 10% de los casos de cáncer de mama.
-
*Características singulares del cáncer de mama familiar:
inicio temprano (premenopáusico) y bilateral*
En general, un cáncer de transmisión hereditaria autosómica
se puede distinguir de un grupo de casos esporádicos en una misma
familia por los dos criterios siguientes: (1) menor edad de aparición
y (2) aparición bilateral o multifocal del cáncer. Algunos
de los síndromes de la familia del cáncer tendrán
también señales físicas características y estigmas
(aunque el cáncer de mama no está entre ellos).
-
Asociado a un mayor riesgo de cáncer de ovario, y posiblemente a
un riesgo aumentado de cáncer de próstata en varones heterocigóticos.
-
Heterogeneidad del locus: un nuevo gen 17q21 (BRCA1
información)
implicado en la reparación del DNA y la segregación de
los cromosomas.
-
Hay un segundo locus en el cromosoma 13 (BRCA2
información),
que codifica también un gen nuevo, probablemente implicado asimismo
en la reparación del DNA. Hay familias con cáncer de mama
familiar que no están ligadas a ninguno de los loci, por lo que
quedan por descubrir más genes BRCA.
-
Lamentablemente, no se ha observado mutación somática de
BRCA1 y BRCA2 en el cáncer de mama esporádico no heredado
(en contraste con otros genes supresores de tumores para otros tipos de
tumores).
-
Los genes BRCA demuestran que la diferencia, en el cáncer hereditario,
entre genes supresores de tumores y genes de reparación del DNA
(ver más abajo) no está tan clara como se creía en
un principio. Por ello, algunos han propuesto nuevas nosologías,
tales como genes "porteros" (gatekeeper) y genes "cuidadores" (caretaker).
Personalmente, el autor encuentra esta división innecesaria, confusa
y fuera de ubicación en la experimentación histórica
y la observación clínica.
-
Del 1 al 2% de los judíos asquenacíes (oriundos de Alemania o de la Europa oriental) tienen mutaciones de la
línea germinal en BRCA1 o BRCA2.
Resumen de genes oncosupresores:
- Identificados como los genes responsables de los síndromes
de tumores humanos con herencia autosómica dominante.
- Recesivos a nivel celular, lo que significa que se requiere la
inactivación de ambos alelos.
- Los casos esporádicos de tumor de línea germinal
muestran a menudo también mutación, especialmente deleción
que produce carencia de heterocigosis.
- Reguladores negativos del crecimiento celular.
3. Genes de reparación del DNA y del ciclo celular
Síndromes autosómicos recesivos de deficiencia en la reparación del DNA
A continuación se relaciona una serie de síndromes infrecuentes de
enfermedad, en la mayoría de los casos cáncer de piel fotosensible
y enfermedad hematológica. Todos ellos están caracterizados
por una deficiencia celular o bioquímica en la reparación
del DNA. Cada enfermedad es heterogénea y se han definido varios "grupos
de complementación" celulares. Se han identificado varios genes
responsables, la mayoría mediante estudios de complementación
en cultivo celular o análisis bioquímico (no de ligamiento).
Como grupo, el estudio de estos genes ha sugerido algunos vínculos
interesantes entre transcripción y reparación del DNA, entre
otras cosas.
-
Síndrome de Bloom: helicasa.
información
-
Xerodermia pigmentaria: diversos factores de trancripción y nucleasas.
información
-
Ataxia telangiectasia: "ATM" - reconoce cromosomas rotos y detiene el
ciclo celular para dar tiempo a que se repare el DNA.
información
-
Anemia de Fanconi
información
-
Síndrome de Cockayne
información
Cáncer de colon hereditario no asociado a poliposis y reparación de errores en el DNA
(HNPCC: hereditary nonpolyposis colon cancer)
información
Síndrome de la familia del cáncer HNPCC-Lynch:
susceptibilidad a numerosos carcinomas, especialmente los de colon (potencialmente,
hasta un 10% de todos los cánceres de colon), otras enfermedades
GI y carcinoma de útero.
-
Se había observado inestabilidad genómica de secuencias repetidas
en tumores de pacientes de familas HNPCC. El patrón de inestabilidad
recuerda al fenotipo de levaduras con deficiente reparación de errores
en el DNA.
-
El gen está fuertemente conservado desde E. coli hasta humanos.
-
Las mutaciones somáticas en genes de reparación de errores
parecen importantes en los casos esporádicos, pues también
se observa inestabilidad genómica de secuencias repetidas en muchos
casos no familiares.
-
Una mutación en un gen de reparación del DNA podría
conducir a una cascada de mutaciones en muchos otros genes, incluyendo
protoncogenes y genes supresores de tumores.
Resumen:
- Los casos infrecuentes de cáncer han enriquecido el conocimiento
de los tumores comunes.
- Los estudios de virus transformantes de RNA condujeron al descubrimiento
de los protoncogenes, que están mutados somáticamente (y
activados de forma dominante) en muchas formas de cáncer.
- Los estudios de cáncer familiar han conducido a la identificación
de los genes supresores de tumores, mostrando también inactivación
(de ambos alelos) en casos esporádicos de tipo similar.
- Los estudios de reparación del DNA, iniciados en E.
coli y levadura, condujeron al descubrimiento de los genes de reparación
de errores en el DNA, implicados en el cáncer familiar con herencia autosómica dominante, así
como en tumores esporádicos.
- El cáncer es, en general, una ruta de múltiples
etapas que requiere la acumulación de mutaciones en varios protoncogenes
o genes supresores de tumores. Este proceso puede potencialmente acelerarse
si se ve afectado un gen de reparación del DNA.